a Division of Paediatric Cardiology, Paediatric Heart Centre, University Children’s Hospital Zurich, Switzerland; b Department of Paediatric Oncology, University Children’s Hospital Zurich, Switzerland; c Institute of Pathology and Molecular Pathology, University Hospital, Zurich, Switzerland; d Department of Paediatric Heart Surgery, Paediatric Heart Centre, University Children’s Hospital Zurich, Switzerland; e Children’s Research Centre Zurich, University Children’s Hospital Zurich
On her way to the supermarket, a 16-year-old teenager collapsed after a sudden stabbing abdominal pain radiating to the back. She was referred to a local hospital unconscious and in cardiogenic shock. Echocardiography revealed a large pericardial effusion; cardiac anatomy and function were normal. Pericardiocentesis yielded 100 ml of blood. With additional volume supply, vital parameters were stabilised. Aortic dissection was suspected and the patient was immediately transferred to our hospital for further treatment.
A thoracic and abdominal computed tomography (CT) scan did not show any aortic dissection and could not identify any other bleeding site. Because of the still ongoing haemorrhage via the pericardial drain an explorative midline sternotomy was performed, which revealed a 3 cm × 5 cm mass situated in the right atrium infiltrating the myocardium. Because of the unknown dignity, the tumour was partially resected and histological results were awaited before planning further best evidence-based treatment.
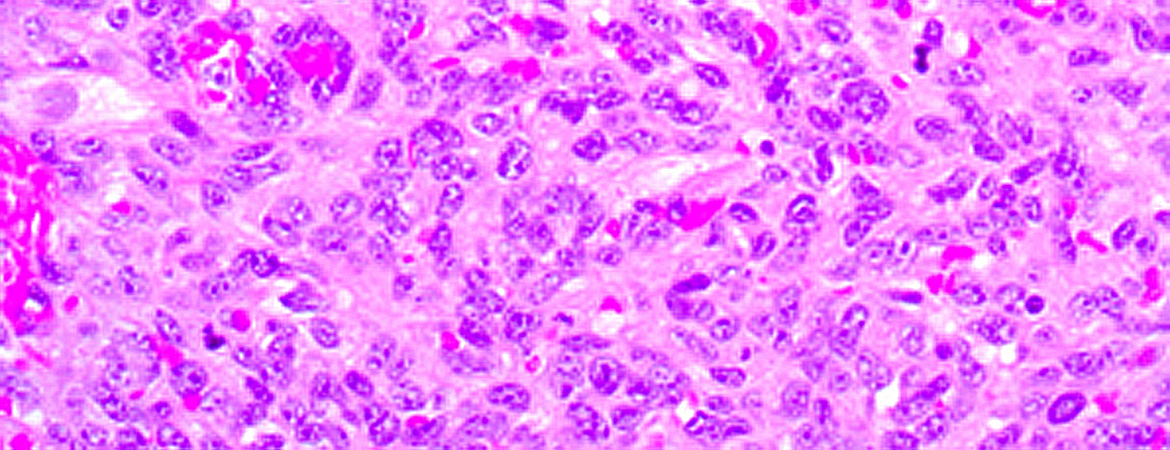
Histological workup of the frozen section showed a malignant mesenchymal tumour, and staining revealed an angiosarcoma (fig. 1). Whole body and cardiac magnetic resonance imaging (MRI) (fig. 2) and the analysis of peritoneal fluid taken during the first operation showed no evidence of a primary extracardiac tumour nor metastases.
Figure 1: (a) Solid tumour with atypical epithelioid and spindle cells and (b) erythroblast transformation specific related gene (ERG) immunohistochemical staining showed diffuse nuclear positivity.
Figure 2: Cardiac magnetic resonance (CMR) cine images (steady state free precession) in an axial (a) and in a short axis (b) view of the residual tumor after first resection.
The mass (**) can be recognised on the back of the right atrial appendage, at the entrance of the superior vena cava. The images also show presence of pericardial effusion (arrows).
LA: left atrium; RA: right atrium; RV: right ventricle; RVOT: right ventricular outflow tract; SVC: superior vena cava.
After interdisciplinary discussion, a further treatment strategy with curative intent was chosen. It consisted of a complete surgical tumour excision followed by chemotherapy.
Cardiac surgery was performed 10 days after the first explorative sternotomy. The whole tumour infiltrating the right atrium could be resected in one piece while the patient was on cardiopulmonary bypass. The right atrial wall was reconstructed using a xenopericardial patch. Resection edges were histologically proven to be tumour free (R0). The patient remained in sinus rhythm and no arrhythmias occurred peri- or postoperatively.
The resected tumour was characterised by a predominantly solid growth pattern with haemorrhagic areas and necrosis. It was composed of epitheloid and spindle cells with marked atypia. The immunophenotype showed an angiomatous differentiation (positive staining for erythroblast transformation specific related gene [ERG]) corresponding to an angiosarcoma (figs 1a and 1b).
Postoperative recovery was uneventful. Chemotherapy following the Cooperative Weichteilsarkom Studiengruppe (CWG) Guidance 2009 “Rhabdomyosarcoma-Like Tumours” was started 9 days after surgery and lasted for 5.5 months (cumulative doses: ifosfamide: 54 g/m2, vincristine 10 mg/m2, doxorubicin: 320 mg/m2, dactinomycin: 6.25 mg/m2). As a result of severe polyneuropathy of the lower extremities vincristine was discontinued after five courses.
Follow-up consisted of regular clinical visits combined with echocardiography, electrocardiogram (ECG), 24h-ECG and cardiac MRI. The patient has remained asymptomatic up to the present (5 years of follow-up). No examinations detected any sign of tumour recurrence or metastasis. The patient completed her education and works now as an office clerk.
Discussion
We present a rare case of a primary cardiac sarcoma successfully treated by a staged approach. Primary cardiac malignancies are extremely rare, with an incidence below 0.007 per 100 000 children, and sarcomas account for about 40% [1, 2]. In the literature, about 180 cases (adults and children) with primary cardiac sarcomas have been described over the last four decades.
The initial presentation of this aggressive tumour is often acute and at an already advanced stage [1, 5] with metastases present in 66–89% of the patients [1, 2]. Tumour location in the right atrium is typical for cardiac sarcomas [3, 5].
In this case, the leading symptoms were related to a massive pericardial effusion. The tumour caused a covered perforation in the right atrial wall, but was not visible in the CT scan – a known difficulty because the tumour mass and the surrounding effusion or myocardial mass may have similar densities. [7] Even MRI could barely depict the tumour (fig. 2). In our patient, the acute bleeding resulted in early detection of the tumour, which was still small and without metastases. In terms of long-term survival, this might have been life-saving.
The prognosis of sarcomas is usually very poor, with a median survival of only 1 month without surgery, and of up to 12 months with surgical resection [1, 6]. Aggressive complete resection in combination with chemotherapy has a better prognosis [1, 3, 4, 8], as was the case for our patient.
A proposed treatment strategy for this kind of tumour is to start chemotherapy before the surgical excision. The aims of this preoperative chemotherapy are to reduce the tumour mass and to treat possible metastasis, since prognosis depends on the following three factors: the presence of metastasis, the side of the tumour (a right-sided tumour being worse) and the ability to achieve an R0 resection [4].
Our patient had initially undergone emergency sternotomy to stop the life-threatening bleeding. Waiting for a first course of chemotherapy would have led to retrosternal adhesions, thus complicating further cardiac surgery. Therefore we decided to perform complete surgical excision first, and start chemotherapy after the total resection of the tumour. The fact that the tumour could be resected in toto may have improved the patient’s prognosis [4, 6, 8].
Neoadjuvant chemotherapy is an important strategy enabling a complete resection by reducing the tumour mass, but was not performed in our patient for the above-mentioned reasons. It is of note that the mainstay of therapy is aggressive surgery, which has more influence on outcome than additional treatments such as chemotherapy and radiotherapy [1, 3, 4, 6, 8]. Radiation can be used as an additional treatment option in selected cases, namely those with microscopic or macroscopic residual disease after surgery [5, 8]. However, the suggested dose for radiation therapy for rhabdomyosarcoma-like tumours needs to be reduced and adjusted to the cardiac location and to patient’s size.
Whether our patient additionally benefited from chemotherapy given according to a paediatric protocol, which was more aggressive than a usual adult protocol, remains unclear.
In cases in which a R0 resection cannot be performed studies are controversial whether heart transplantation is a valuable treatment option [4]. For staging and follow up MRI and positron emission tomography (PET)-CT are recommended modalities.
The course of our patient, who presented in an acute state, without metastases and with a successful R0 resection, is extremely rare [1]. The long, uneventful survival of 5 years is extraordinary. Early diagnosis and aggressive resection are key points when facing a patient with cardiac sarcoma.
Disclosure statement
No financial support and no other potential conflict of interest relevant to this article was reported.
Correspondence
Correspondence: Anna Cavigelli-Brunner, MD Division of Paediatric Cardiology Paediatric Heart Centre University Children’s Hospital Zurich Steinwiesstr. 75 CH-8032 Zurich anna.cavigelli[at]kispi.uzh.ch
References
1 Hudzik B, Miszalski-Jamka K, Glowacki J, Lekston A, Gierlotka M, et al. Malignant tumors of the heart. Cancer Epidemiol. 2015;39(5):665–72. doi: 10.1016/j.canep.2015.07.007. Epub 2015 Aug 1.
2 Davis JS, Allan BJ, Perez EA, Neville HL, Sola JE. Primary pediatric cardiac malignancies: the SEER experience Pediatr Surg Int. 2013;29(5):425–9. doi: 10.1007/s00383-013-3261-4. Epub 2013 Jan 29.
3 Ramlawi B, Leja M, Walid K, Saleh A, Al Jabbari O, Benjamin R, et al. Surgical Treatment of Primary Cardiac Sarcomas: Review of a Single-Institution Experience. Ann Thorac Surg. 2016;101:698–702.
4 Habertheuer A, Laufer G, Wiedemann D, Andreas M, Ehrlich M, et al. Primarycardiac tumors on the verge of oblivion: a European experience over 15 years. J Cardiothorac Surg. 2015;10:56.
5 Look Hong NJ, Pandalai PK, Hornick JL, Shekar PS, Harmon DC, Chen YL, et al. Cardiac angiosarcoma management and outcomes: 20-year single institution experience. Ann Surg Oncol. 2012;19:2707–15.
6 ElBardissi AW, Dearani JA, Daly RC, Mullany CJ, Orszulak TA, et al. Survival After Resection of Primary Cardiac Tumors – A 48-Year Experience. Circulation. 2008;118:S7–S15.
7 Burnside N, Jeganathan R. Cardiac sarcoma causing mechanical tamponade: a radiological dilemma! Eur Heart J. 2015;36(48):3459.
8 Abu Saleh WK, Ramlawi B, Shapira OM, Al Jabbari O, Ravi V, et al. Improved Outcomes With the Evolution of a Neoadjuvant Chemotherapy Approach to Right Heart Sarcoma. Ann Thorac Surg. 2017;104:90–7.