a Department of Cardiology, University Heart Centre, University Hospital Zurich, Switzerland; b Cardio-surgical Intensive Care Unit, Institute of Anaesthesiology, University Hospital Zurich, Switzerland
Heart failure is a clinical syndrome induced by structural and/or functional cardiac abnormalities, resulting in reduced cardiac output and/or elevated intracardiac end-diastolic pressures and causing symptoms, often accompanied by typical physical signs [1]. The number of subjects living with heart failure has increased over recent decades because of demographic changes and, at least in part, because of the success of modern cardiology, which achieved a marked reduction in short-term mortality of several acute conditions (e.g., myocardial infarction, arrhythmia, congenital heart disease) and, by combining several drug classes and devices, improved long-term survival of patients with reduced left-ventricular ejection fraction. As a consequence, heart failure has become a major public health problem, affecting 2% of the adult population in developed countries, and the number of hospital admissions related to heart failure has tripled over the last three decades [2].
Acute heart failure (AHF) is defined as new-onset or worsening of symptoms and signs of heart failure and is the most frequent cause of unplanned hospital admission in patients aged 65 years or older. Despite major achievements in the treatment of chronic heart failure, outcomes of AHF remain poor, with high mortality and readmission rates. In this context, there is an unmet need for individualisation of AHF treatment according to the underlying pathophysiological mechanisms and for optimisation of transitions of care to, hopefully, improve patient outcome.
Congestion and the pathophysiology of acute heart failure
Heart failure was historically described with the haemodynamic model of a pump failure causing downstream hypoperfusion (forward failure) and upstream congestion (backward failure). Over recent decades, a more complex network of interactions has been added to this model for explaining the pathophysiology of heart failure. AHF may arise in patients without previous history of symptomatic heart failure (de novo AHF), mostly secondarily to an acute deterioration of the cardiac function (e.g., myocardial infarction, severe myocarditis, acute valve regurgitation) causing relevant haemodynamic alterations [3]. More frequently, AHF consists of acute decompensations of chronic heart failure (ADHF) and is caused by progressive congestion that may be precipitated by several factors [4].
Cardiac dysfunction induces the activation of several neuro-humoral pathways (e.g., the sympathetic nervous system, the renin-angiotensin-aldosterone system and the arginine-vasopressin system) to counter the negative haemodynamic effects of heart failure [5]. Neuro-humoral activation leads to impaired sodium excretion through the kidneys, which results in sodium and, secondarily, fluid accumulation [6]. Moreover, persistent neuro-humoral activation induces maladaptive processes resulting in detrimental ventricular remodelling and organ dysfunction.
The congestive cascade includes a subclinical stage (haemodynamic congestion), defined as increase in cardiac filling and venous pressures, followed by redistribution of fluids into the lungs and visceral organs (organ congestion), and finally to overt symptoms and signs of volume overload (clinical congestion) [7]. Although clinical and organ congestion usually follow haemodynamic congestion, the correlation between hydrostatic pressure and oedema formation is poor. Indeed, chronic sodium accumulation in heart failure impairs the function of the interstitial glycosaminoglycan network, reducing its capacity to buffer additional sodium and maintain low interstitial compliance [8]. Consequently, interstitial oedema formation may occur even in the presence of mildly elevated hydrostatic pressures.
Many AHF patients display only a minor increase in body weight before hospital admission and congestion is precipitated mainly by a sudden fluid redistribution rather than fluid accumulation. Sympathetic activation has been shown to induce transient vasoconstriction leading to rapid volume displacement from the peripheral and splanchnic venous systems to the pulmonary circulation [9]. A mismatch in the ventricular-vascular coupling relationship, with increased afterload and decreased venous capacitance (increased preload), is the primary alteration in hypertensive AHF [10, 11]. Fluid accumulation and fluid redistribution both promote systemic congestion in AHF, but their contribution varies according to different clinical situations. Whereas fluid redistribution might be the predominant mechanism in acute de novo forms and in ADHF with preserved ejection fraction, fluid accumulation might be more common in ADHF with reduced ejection fraction.
Recent data suggested that venous congestion is not simply an epiphenomenon secondary to cardiac dysfunction, but rather plays an active detrimental role, inducing pro-oxidant, pro-inflammatory, and haemodynamic stimuli that contribute to the pathophysiology of AHF [12, 13].
Furthermore, systemic congestion has been recognised as a central feature in AHF, since it is the main determinant of the clinical presentation and significantly contributes to organ dysfunction in AHF. Left-sided congestion may cause dyspnoea, orthopnoea, bendopnoea (shortness of breath when leaning forward), paroxysmal nocturnal dyspnoea, cough, tachypnoea, hypoxia and pathological lung auscultation. Right-sided congestion may cause pleural effusions, jugular vein distension or positive hepatojugular reflux, ascites, hepatomegaly, icterus, abdominal pain, nausea, vomiting and decreased urine output. Peripheral oedema is not specific for right heart failure, since it can be promoted by neuro-humoral activation and fluid overload induced by left and/or right heart failure. Of note, leg oedema can be absent in the early phase of AHF. Historically, renal and hepatic dysfunctions in heart failure have been considered as the consequence of visceral hypoperfusion, but more recent data showed that central venous pressure (reflecting venous congestion) was the strongest haemodynamic determinant of renal and hepatic dysfunction in AHF [14–16]. Symptoms and signs of hypoperfusion are much less common and indicate severity. These include hypotension, tachycardia, weak pulse, mental confusion, anxiety, fatigue, dyspnoea/tachypnoea (due to cerebral hypoxemia and/or and metabolic acidosis), cold sweaty extremities, decreased urine output, and angina due to myocardial ischaemia.
Diagnosis and initial treatment
The basis for the correct management of AHF patients depends on the correct diagnosis of the cardiac disease, the pathophysiology at play and comorbidities (table 1).
Table 1: Principles for the correct management of patients with acute heart failure.
Capture comorbidities / relative contraindications to diagnostics or treatment
Severe renal failure Active bleeding / bleeding risk Arterial hypertension Diabetes mellitus COPD, smoking Allergy to contrast media Pregnancy
Consider iatrogenic harm
Hyopkalaemia after diuretics Hypotension after ACE inhibitors Bradycardia / negative inotropy after beta-blockers Hyperkalaemia after mineralocorticoid receptor antagonists
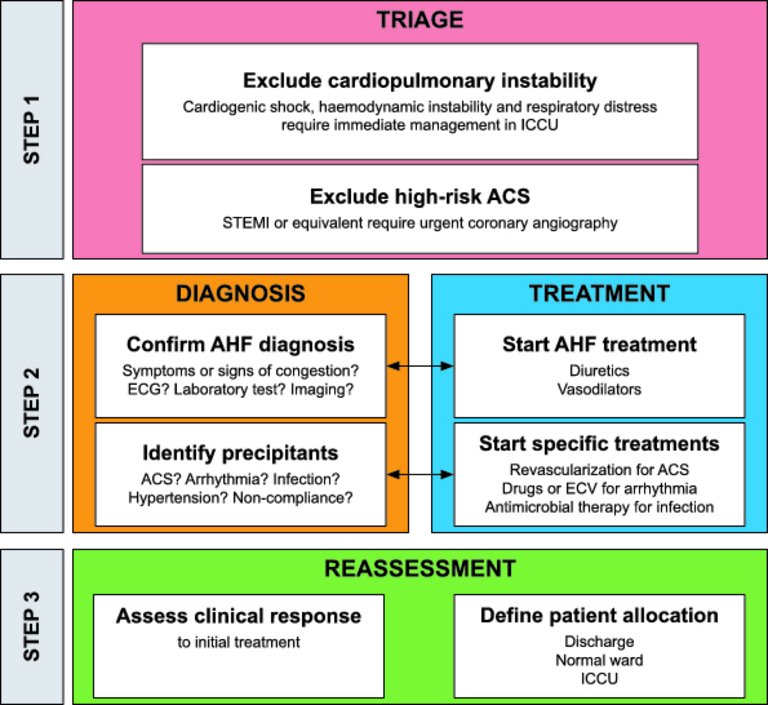
The treatment consists of three steps: triage, confirmation of diagnosis and initiation of treatment, and reassessment (fig. 1). Since AHF is a life-threatening condition, diagnosis and initiation of treatment go hand in hand and should be initiated within 30 to 60 minutes after hospital admission [17].
Figure 1: Initial management of acute heart failure.
ACS = acute coronary syndrome; AHF = acute heart failure; ECV = electrical cardioversion; ICCU = intensive cardiac care unit; STEMI = ST-elevation myocardial infarction. Modified from: Arrigo M, Parissis JT, Akiyama E, Mebazaa A. Understanding acute heart failure: pathophysiology and diagnosis. Eur Heart J Suppl. 2016;18(suppl G):G11–8, reprinted with permission of Oxford University Press.
During the first step (triage), patients with cardiogenic shock, respiratory insufficiency, myocardial ischaemia and/or arrhythmia (table 2) should be identified and receive an appropriate level of monitoring and specific treatments (e.g., mechanical ventilation, pharmacological haemodynamic support). Since congestion is the typical feature of AHF, history and physical examination should primarily focus on the presence of congestion, which would support the diagnosis of AHF. However, given the limited sensitivity and specificity of symptoms and signs of AHF, the clinical evaluation should integrate information from additional tests. According to the presence of clinical symptoms or signs of organ congestion (“wet” vs “dry”) and/or peripheral hypoperfusion (“cold” vs “warm”), patients may be classified in four groups (fig. 2). About two out of three AHF patients are classified “warm-wet” (well perfused but congested), about one in four are “cold-wet” (hypoperfused and congested) and only a minority are “cold-dry” (hypoperfused but not congested). Hypoperfusion defines cardiogenic shock, which accounts for less than 10% of AHF cases but is associated with in-hospital mortality rates of up to 50% [20]. The fourth group “warm-dry” represents the compensated (well-perfused, decongested) status. This classification may help to guide initial therapy (see below). During triage, patients with high-risk acute coronary syndrome (e.g., ST-elevation myocardial infarction or equivalent) should be recognised and rapidly transferred to the catheterisation laboratory to receive coronary angiography with percutaneous revascularisation.
Figure 2: Haemodynamic profiles of acute heart failure according to the presence of congestion and hypoperfusion. Modified from Forrester et al. [18, 19].
Table 2: Medical emergencies in patients with acute heart failure.
Ventricular tachycardia Atrial fibrillation and signs of AHF Bradycardia, AV block II or III
The second step consists of confirmation of diagnosis and initiation of treatment. Natriuretic peptides, either B-type NP (BNP), amino-terminal pro-B-type NP (NT-proBNP) or mid-regional pro-atrial NP (MR-proANP) should be measured in patients with suspected AHF, since they show high accuracy and excellent negative predictive value in differentiating AHF from non-cardiac causes of acute dyspnoea. Low circulating natriuretic peptides (thresholds: BNP <100 pg/ml, NT-proBNP <300 pg/ml, MR-proANP <120 pmol/l) make the diagnosis of AHF unlikely. However, elevated concentrations of natriuretic peptides do not automatically confirm the diagnosis of AHF, as they may also be secondary to a wide variety of cardiac and non-cardiac causes (e.g., atrial fibrillation, renal failure, infections) [21]. On the other hand, circulating natriuretic peptides may be falsely low in obese patients and in those with flash pulmonary oedema. The electrocardiogram is rarely normal in AHF patients, but may be helpful for identifying potential precipitants of AHF (e.g., arrhythmia, ischaemia) and to exclude ST-elevation myocardial infarction requiring immediate revascularisation. Thoracic ultrasound and chest X-ray may both be useful to assess the presence of pulmonary congestion. Systemic congestion may be assessed by means of sonographic measurement of the inferior vena cava diameter and collapsibility. In the presence of cardiogenic shock or suspicion of acute life-threatening structural or functional cardiac abnormalities (mechanical complications, acute valve regurgitation), immediate echocardiography should be performed [22]. Early echocardiography (preferably within 24 hours from admission) should be considered in all patients with de novo AHF and in those with unknown cardiac function.
The initial treatment of “wet” patients with evidence of congestion frequently includes diuretics and vasodilators [23]. Whereas diuretics are mainly used in presence of fluid overload, vasodilators are administered to reduce filling pressures in the presence of fluid redistribution and preserved systolic blood pressure (>110 mm Hg, more cautiously and in experienced hands between 90 and 110 mm Hg). Decongestive therapy should be started as soon as possible, titrated according to the clinical response, and maintained until euvolaemia is achieved [24]. In patients with severe pulmonary oedema, high-flow oxygen therapy, noninvasive ventilation or mechanical ventilation are required to ensure oxygenation. Patients with de novo AHF with reduced left-ventricular ejection fraction should be started with oral disease-modifying heart failure treatment (angiotensin converting-enzyme inhibitors, beta-blockers and mineralocorticoid receptor antagonists) after initial decongestion, optimally before hospital discharge (see below). Patients presenting with ADHF should be maintained on oral disease-modifying heart failure treatment whenever possible.
Notably, the use of inotropes should be restricted to “cold” patients in cardiogenic shock with impaired myocardial contractility, since their often inappropriate use is associated with increased morbidity and mortality [25]. In cases of persistent haemodynamic instability despite escalating doses of inotropes, mechanical circulatory support (e.g., venoarterial extracorporeal membrane oxygenation, va-ECMO) should be considered at an early stage to restore organ perfusion [26].
As mentioned above, AHF may be precipitated by several factors, which may coexist (e.g., myocardial ischemia, arrhythmias, infections, uncontrolled hypertension, noncompliance with medical prescriptions). The identification of precipitants during early management aims at detecting treatable causes and provides prognostic information. Indeed, according to current knowledge on the pathophysiology of AHF, the initial management should include, in addition to decongestive therapy (e.g., vasodilators and/or diuretics), specific treatments directed towards the underlying causes of AHF. In particular, early coronary angiography with revascularisation is recommended in AHF precipitated by acute coronary syndrome, antiarrhythmic treatment and/or electrical cardioversion are recommended in AHF precipitated by arrhythmia, and rapid initiation of antimicrobial therapy is recommended for AHF precipitated by infections. Sometimes percutaneous or surgical treatment of structural heart disease is required to achieve durable stabilisation. Furthermore, identification of precipitants of AHF may allow risk stratification of patients with AHF. Indeed, AHF precipitated by acute coronary syndrome or infection is associated with poorer outcomes, whereas outcomes tend to be better in AHF precipitated by atrial fibrillation or uncontrolled hypertension [4, 27, 28].
The third step of the initial management consists in reassessment of the clinical response to initial treatment and decision about patient allocation. The level of care (discharge, observation, ward, telemetry, and intensive or intermediate care unit) should be based on history (symptom severity, precipitating factors), physical examination (haemodynamic and respiratory status, degree of congestion) and biomarkers (natriuretic peptides, troponin, renal function, serum lactate). Most patients require hospital admission, about half of them to intensive or intermediate care units. Low-risk patients with a good response to initial therapy may be considered for early discharge.
Hospital discharge and the vulnerable phase
The decision about the optimal time-point for hospital discharge of hospitalised patients with AHF may be challenging. Indeed, the early post-discharge period immediately following a hospitalisation for AHF is characterised by relevant short-term mortality and frequent hospital readmissions (fig. 3), causing a significant burden in healthcare expenditures [30].
Figure 3: The “vulnerable phase” after discharge for acute heart failure. The first 3 months after hospital discharge for AHF are characterised by high rate of adverse events (death and readmission). This phase is commonly defined as “vulnerable phase”. Modified from Greene et al. [29].
The causes of this “vulnerable phase”, which usually lasts 2 to 3 months, remain controversial [29]. The combination of pathophysiological derangements (e.g., persistent congestion, insufficient neuro-humoral blockade) and healthcare system problems (e.g., lack of follow-up) seems to contribute to the poor outcome.
In this context, clinicians need to balance between the risk of adverse outcome, healthcare costs and patient preferences in order to determine the optimal length of hospital stay.
Some studies suggested persistent haemodynamic congestion at hospital discharge as a critical factor in the pathophysiology underlying the high rates of death and readmission during the vulnerable phase [31]. Indeed, although most patients admitted with AHF report improvement in symptoms during the hospital stay, circulating natriuretic peptides may still be markedly elevated at discharge. This discordance between few symptoms of congestion and high natriuretic peptide concentrations suggests persistent haemodynamic congestion at discharge. Some studies reported an association between predischarge levels of natriuretic peptides and the risk of death or readmission during the vulnerable phase [32]. Based on these observations, the titration of the decongestive therapy based only on symptoms and signs of congestion may be insufficient and should include additional parameters (e.g., biomarkers, echocardiography) [33, 34].
Underutilisation of disease-modifying HF therapies (e.g., beta-blockers, renin-angiotensin system inhibitors [RASIs] and mineralocorticoid receptor antagonists [MRAs]) may further promote adverse events after hospital discharge. A recent meta-analysis showed detrimental effects on short-term mortality and readmission associated with beta-blocker discontinuation during AHF hospitalisation [35]. Despite lacking data on RASIs and MRAs, the current European Society for Cardiology guidelines recommend initiation or continuation of disease-modifying HF therapies during hospitalisation in all AHF patients with reduced ejection fraction, unless contraindicated [1]. Very recently, a large, propensity score-matched, cohort study showed an association between beta-blocker or RASI treatment at hospital discharge and a 40 to 50% relative risk reduction in 90-day mortality, and an additional 25 to 50% relative risk reduction associated with combined treatment with a beta-blocker and RASI at hospital discharge compared with a beta-blocker or RASI alone [36]. The early benefits were present with both reduced and preserved ejection fraction and persisted at 1-year follow-up. In the same study, no early benefit of early administration of MRAs was found. In patients intolerant to beta-blockers, early initiation of ivabradine might be considered to reduce readmissions during the vulnerable phase [37]. In light of these data, continuation or initiation of disease-modifying heart failure drugs (e.g., beta-blocker, RASI) before hospital discharge in all AHF patients, unless clearly contraindicated, seems reasonable.
A further critical point to consider before hospital discharge is whether the precipitating factors of AHF have been adequately treated and resolved. This includes revascularisation for myocardial ischaemia, antiarrhythmic therapy for arrhythmias, antimicrobial therapy for infection, antihypertensive therapy for hypertension and patient education for malcompliance. Furthermore, clinicians should take care of a delayed peak in the risk of death in AHF precipitated by infection, occurring at weeks 3 to 4 after admission (fig. 4) [4].
Figure 4: Time-course of risk of death according to precipitating factors and comorbidities. The weekly risk of death associated with the presence of selected precipitating factors of AHF (red, acute coronary syndrome; green, infection; blue, atrial fibrillation) compared with absence of identified precipitating factors. The weekly risk of death associated with the presence of multiple co-morbidities (AHEAD score ≥4 points) compared with score <4 points is depicted in grey. Modified from Arrigo M, Gayat E, Parenica J, Ishihara S, Zhang J, Choi DJ, et al.; GREAT Network. Precipitating factors and 90-day outcome of acute heart failure: a report from the intercontinental GREAT registry. Eur J Heart Fail. 2017;19(2):201–8, reprint with permission. ACS = acute coronary syndrome; AHF = acute heart failure
Patient education, home-based collection of subjective and objective data (symptoms, body weight, blood pressure) and early contact with healthcare providers have been proposed to reduce readmission rates, but with inconsistent results. Several strategies to achieve early detection of congestion, including intrathoracic impedance monitoring and implantable haemodynamic monitoring (e.g., the CardioMEMS HF system), showed promising results in the trials, but their wide applicability remains questionable, mainly because of uncertainty about cost effectiveness [38, 39].
Optimally, all patients with AHF should benefit from inclusion in an early, comprehensive, post-discharge care programme. If this is not feasible because of limited resources, it should be restricted to high-risk patients (e.g., elevated natriuretic peptides, abnormal systolic blood pressure, persistent hyponatraemia, recurrent readmissions) [29]. In our centre, we recommend an early visit to the general practitioner within 1 week after discharge and we schedule a visit to the HF outpatient clinic within 2 to 3 weeks after discharge. During these early outpatient visits, assessment and optimisation of the volume status (including comparison of natriuretic peptide concentrations with predischarge values) and up-titration of disease-modifying HF treatment should be performed.
Key points
– Acute heart failure is a life-threatening condition requiring immediate diagnosis and initiation of treatment.
– Systemic congestion, promoted by the activation of several pathophysiological mechanisms, is the central feature of acute heart failure and causes the typical symptoms and leads to organ dysfunction.
– Cardiogenic shock is the most severe form of acute heart failure and is defined by the presence of reduced cardiac output and end-organ hypoperfusion.
– Decongestive therapy is the mainstay in the initial treatment of acute heart failure with congestion. Inotropes are used in cardiogenic shock patients with reduced myocardial contractility to restore end-organ perfusion.
– The first 3 months after an acute heart failure episode – the vulnerable phase – are characterised by high readmission and mortality rates. Optimal patient management before and after hospital discharge is crucial to improve patient outcome.
Disclosure statement
MA reports speaker fees from Orion Pharma, outside the submitted work. AR reports speaker fees from OrphaSwiss GmbH and Amomed Pharma GmbH, outside the submitted work.
Correspondence
Correspondence: Mattia Arrigo, MD Deputy Head Acute Cardiology Department of Cardiology University Heart Centre University Hospital Zurich Rämistrasse 100 CH-8091 Zurich mattia.arrigo[at]usz.ch
References
1 Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al.; Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–200. doi:https://doi.org/10.1093/eurheartj/ehw128.
3 Rudiger A, Harjola V-P, Müller A, Mattila E, Säila P, Nieminen M, et al. Acute heart failure: clinical presentation, one-year mortality and prognostic factors. Eur J Heart Fail. 2005;7(4):662–70. doi:https://doi.org/10.1016/j.ejheart.2005.01.014.
4 Arrigo M, Gayat E, Parenica J, Ishihara S, Zhang J, Choi DJ, et al.; GREAT Network. Precipitating factors and 90-day outcome of acute heart failure: a report from the intercontinental GREAT registry. Eur J Heart Fail. 2017;19(2):201–8. doi:https://doi.org/10.1002/ejhf.682.
5 Arrigo M, Parissis JT, Akiyama E, Mebazaa A. Understanding acute heart failure: pathophysiology and diagnosis. Eur Heart J Suppl. 2016;18(suppl G):G11–8. doi:https://doi.org/10.1093/eurheartj/suw044.
6 McKie PM, Schirger JA, Costello-Boerrigter LC, Benike SL, Harstad LK, Bailey KR, et al. Impaired natriuretic and renal endocrine response to acute volume expansion in pre-clinical systolic and diastolic dysfunction. J Am Coll Cardiol. 2011;58(20):2095–103. doi:https://doi.org/10.1016/j.jacc.2011.07.042.
7 Zile MR, Bennett TD, St John Sutton M, Cho YK, Adamson PB, Aaron MF, et al. Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circulation. 2008;118(14):1433–41. doi:https://doi.org/10.1161/CIRCULATIONAHA.108.783910.
8 Nijst P, Verbrugge FH, Grieten L, Dupont M, Steels P, Tang WH, et al. The pathophysiological role of interstitial sodium in heart failure. J Am Coll Cardiol. 2015;65(4):378–88. doi:https://doi.org/10.1016/j.jacc.2014.11.025.
9 Cotter G, Metra M, Milo-Cotter O, Dittrich HC, Gheorghiade M. Fluid overload in acute heart failure – re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail. 2008;10(2):165–9. doi:https://doi.org/10.1016/j.ejheart.2008.01.007.
10 Gandhi SK, Powers JC, Nomeir AM, Fowle K, Kitzman DW, Rankin KM, et al. The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med. 2001;344(1):17–22. doi:https://doi.org/10.1056/NEJM200101043440103.
11 Viau DM, Sala-Mercado JA, Spranger MD, O’Leary DS, Levy PD. The pathophysiology of hypertensive acute heart failure. Heart. 2015;101(23):1861–7. doi:https://doi.org/10.1136/heartjnl-2015-307461.
12 Colombo PC, Onat D, Harxhi A, Demmer RT, Hayashi Y, Jelic S, et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur Heart J. 2014;35(7):448–54. doi:https://doi.org/10.1093/eurheartj/eht456.
13 Harjola V-P, Mullens W, Banaszewski M, Bauersachs J, Brunner-La Rocca HP, Chioncel O, et al. Organ dysfunction, injury and failure in acute heart failure: from pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2017;19(7):821–36. doi:https://doi.org/10.1002/ejhf.872.
14 Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589–96. doi:https://doi.org/10.1016/j.jacc.2008.05.068.
15 Poelzl G, Ess M, Mussner-Seeber C, Pachinger O, Frick M, Ulmer H. Liver dysfunction in chronic heart failure: prevalence, characteristics and prognostic significance. Eur J Clin Invest. 2012;42(2):153–63. doi:https://doi.org/10.1111/j.1365-2362.2011.02573.x.
16 Ishihara S, Gayat E, Sato N, Arrigo M, Laribi S, Legrand M, et al. Similar hemodynamic decongestion with vasodilators and inotropes: systematic review, meta-analysis, and meta-regression of 35 studies on acute heart failure. Clin Res Cardiol. 2016;105(12):971–80. doi:https://doi.org/10.1007/s00392-016-1009-6.
17 Mebazaa A, Yilmaz MB, Levy P, Ponikowski P, Peacock WF, Laribi S, et al. Recommendations on pre-hospital & early hospital management of acute heart failure: a consensus paper from the Heart Failure Association of the European Society of Cardiology, the European Society of Emergency Medicine and the Society of Academic Emergency Medicine. Eur J Heart Fail. 2015;17(6):544–58. doi:https://doi.org/10.1002/ejhf.289.
18 Forrester JS, Diamond G, Chatterjee K, Swan HJ. Medical therapy of acute myocardial infarction by application of hemodynamic subsets (first of two parts). N Engl J Med. 1976;295(24):1356–62. doi:https://doi.org/10.1056/NEJM197612092952406.
19 Forrester JS, Diamond GA, Swan HJ. Correlative classification of clinical and hemodynamic function after acute myocardial infarction. Am J Cardiol. 1977;39(2):137–45. doi:https://doi.org/10.1016/S0002-9149(77)80182-3.
20 Harjola V-P, Lassus J, Sionis A, Køber L, Tarvasmäki T, Spinar J, et al.; CardShock Study Investigators; GREAT network. Clinical picture and risk prediction of short-term mortality in cardiogenic shock. Eur J Heart Fail. 2015;17(5):501–9. doi:https://doi.org/10.1002/ejhf.260.
21 Rudiger A, Gasser S, Fischler M, Hornemann T, von Eckardstein A, Maggiorini M. Comparable increase of B-type natriuretic peptide and amino-terminal pro-B-type natriuretic peptide levels in patients with severe sepsis, septic shock, and acute heart failure. Crit Care Med. 2006;34(8):2140–4. doi:https://doi.org/10.1097/01.CCM.0000229144.97624.90.
22 Mebazaa A, Tolppanen H, Mueller C, Lassus J, DiSomma S, Baksyte G, et al. Acute heart failure and cardiogenic shock: a multidisciplinary practical guidance. Intensive Care Med. 2016;42(2):147–63. doi:https://doi.org/10.1007/s00134-015-4041-5.
23 Cotter G, Metzkor E, Kaluski E, Faigenberg Z, Miller R, Simovitz A, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351(9100):389–93. doi:https://doi.org/10.1016/S0140-6736(97)08417-1.
24 Matsue Y, Damman K, Voors AA, Kagiyama N, Yamaguchi T, Kuroda S, et al. Time-to-Furosemide Treatment and Mortality in Patients Hospitalized With Acute Heart Failure. J Am Coll Cardiol. 2017;69(25):3042–51. doi:https://doi.org/10.1016/j.jacc.2017.04.042.
25 Mebazaa A, Parissis J, Porcher R, Gayat E, Nikolaou M, Boas FV, et al. Short-term survival by treatment among patients hospitalized with acute heart failure: the global ALARM-HF registry using propensity scoring methods. Intensive Care Med. 2011;37(2):290–301. doi:https://doi.org/10.1007/s00134-010-2073-4.
26 Wilhelm MJ. Extracorporeal membrane oxygen ation for acute cardiogenic shock. Cardiovasc Med. 2016;19(2):39–43. doi:https://doi.org/10.4414/cvm.2016.00393.
27 Arrigo M, Tolppanen H, Sadoune M, Feliot E, Teixeira A, Laribi S, et al.; GREAT Network. Effect of precipitating factors of acute heart failure on readmission and long-term mortality. ESC Heart Fail. 2016;3(2):115–21. doi:https://doi.org/10.1002/ehf2.12083.
28 Rudiger A, Streit M, Businger F, Schmid ER, Follath F, Maggiorini M. The impact of infections on critically ill acute heart failure patients: an observational study. Swiss Med Wkly. 2010;140:w13125.
29 Greene SJ, Fonarow GC, Vaduganathan M, Khan SS, Butler J, Gheorghiade M. The vulnerable phase after hospitalization for heart failure. Nat Rev Cardiol. 2015;12(4):220–9. doi:https://doi.org/10.1038/nrcardio.2015.14.
30 Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63(12):1123–33. doi:https://doi.org/10.1016/j.jacc.2013.11.053.
31 Ambrosy AP, Pang PS, Khan S, Konstam MA, Fonarow GC, Traver B, et al.; EVEREST Trial Investigators. Clinical course and predictive value of congestion during hospitalization in patients admitted for worsening signs and symptoms of heart failure with reduced ejection fraction: findings from the EVEREST trial. Eur Heart J. 2013;34(11):835–43. doi:https://doi.org/10.1093/eurheartj/ehs444.
32 Logeart D, Thabut G, Jourdain P, Chavelas C, Beyne P, Beauvais F, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol. 2004;43(4):635–41. doi:https://doi.org/10.1016/j.jacc.2003.09.044.
33 Arrigo M, Truong QA, Onat D, Szymonifka J, Gayat E, Tolppanen H, et al. Soluble CD146 Is a Novel Marker of Systemic Congestion in Heart Failure Patients: An Experimental Mechanistic and Transcardiac Clinical Study. Clin Chem. 2017;63(1):386–93. doi:https://doi.org/10.1373/clinchem.2016.260471.
34 Kubena P, Arrigo M, Parenica J, Gayat E, Sadoune M, Ganovska E, et al.; GREAT Network. Plasma Levels of Soluble CD146 Reflect the Severity of Pulmonary Congestion Better Than Brain Natriuretic Peptide in Acute Coronary Syndrome. Ann Lab Med. 2016;36(4):300–5. doi:https://doi.org/10.3343/alm.2016.36.4.300.
35 Prins KW, Neill JM, Tyler JO, Eckman PM, Duval S. Effects of Beta-Blocker Withdrawal in Acute Decompensated Heart Failure: A Systematic Review and Meta-Analysis. JACC Heart Fail. 2015;3(8):647–53. doi:https://doi.org/10.1016/j.jchf.2015.03.008.
36 Gayat E, Arrigo M, Littnerova S, Sato N, Parenica J, Ishihara S, et al.; GREAT Network. Heart failure oral therapies at discharge are associated with better outcome in acute heart failure: a propensity-score matched study. Eur J Heart Fail. 2017;18:613.
37 Komajda M, Tavazzi L, Swedberg K, Böhm M, Borer JS, Moyne A, et al.; SHIFT Investigators. Chronic exposure to ivabradine reduces readmissions in the vulnerable phase after hospitalization for worsening systolic heart failure: a post-hoc analysis of SHIFT. Eur J Heart Fail. 2016;18(9):1182–9. doi:https://doi.org/10.1002/ejhf.582.
38 Hindricks G, Taborsky M, Glikson M, Heinrich U, Schumacher B, Katz A, et al.; IN-TIME study group*. Implant-based multiparameter telemonitoring of patients with heart failure (IN-TIME): a randomised controlled trial. Lancet. 2014;384(9943):583–90. doi:https://doi.org/10.1016/S0140-6736(14)61176-4.
39 Abraham WT, Adamson PB, Bourge RC, Aaron MF, Costanzo MR, Stevenson LW, et al.; CHAMPION Trial Study Group. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet. 2011;377(9766):658–66. doi:https://doi.org/10.1016/S0140-6736(11)60101-3.