First historical descriptions of arrhythmogenic cardiomyopathy (ACM) were made in the 18th century. Initially it was thought to be an embryological malformation, but in recent years it was shown that the disease is a genetically determined progressive myocardial disease leading to fibro-fatty infiltration [1]. Therefore, in 1995 it was classified as a primary cardiomyopathy by the World Health Organization [2]. In its most typical form, the right ventricle (RV) is primarily affected, which is referred to as arrhythmogenic right ventricular cardiomyopathy (ARVC). In Europe, the disease is more common in males (2–3:1), and usually manifests after puberty. The general prevalence is around 1:2000–1:5000, which makes ACM a rare disease. Nonetheless, it is one of the leading causes of sudden cardiac death (SCD), particularly in young athletes ≤35 years of age [3].
Pathogenesis
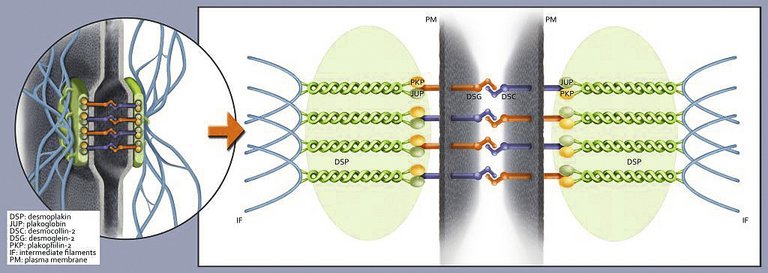
ACM is usually inherited as an autosomal dominant trait with incomplete penetrance. There are also very rare recessive forms, which can cause severe phenotypic expression, such as Naxos disease which includes skin and hair involvement. Moreover, it has been shown that in up to 18% of patients with ACM, compound or digenic heterozygosity is present, meaning that both alleles of a gene, or different genes may be affected. To date, >13 different genetic loci have been reported to be associated with ACM and the most common genes encode for desmosomal proteins [4]. Desmosomes provide cardiomyocyte-to-cardiomyocyte adhesion. In about 80% of mutation positive patients, mutations are found in the desmosomal genes plakophilin-2 (PKP2), desmoglein-2 (DSG2), and desmoplakin (DSP) (fig. 1) [5, 6]. A defective desmosome can result in intercalated disk remodelling, which alters not only mechanical stability, but also electrical coupling between cells, and intra/intercellular signal transduction, affecting apoptosis and lipid metabolism and ultimately leading to fibro-fatty tissue replacement [7–9]. Myocardial remodelling starts in the subepicardial layers and may become transmural later on.
Figure 1: Illustration of the intercalated disc with the desmosome as its key component (left), an important cellular structure providing cell-cell adhesion and mechanical integrity within the heart muscle. The enlarged sector (right) schematizes the desmosomal network and its major components. ARVC indicates arrhythmogenic right ventricular cardiomyopathy.
From: Saguner AM, Duru F, Brunckhorst CB. Arrhythmogenic right ventricular cardiomyopathy. Circulation. 2013;128:1381–6, reprinted with permission. Promotional and commercial use of the material in print, digital or mobile device format is prohibited without the permission from the publisher Wolters Kluwer. Please contact healthpermissions@wolterscluwer.com for further information.
Some mutations, for example in DSP, certain mutations in the non-desmosomal transmembrane protein 43 (TMEM43), or compound/digenic mutations have been associated with more aggressive phenotypes and, therefore, can be considered as risk factors of heart failure and SCD. Nonetheless, further studies with larger patient cohorts need to be conducted in order to improve our understanding of genotype-phenotype correlations in ACM. Recent methods of genetic testing such as next generation sequencing are able to process large amounts of genetic data in a short time. Nevertheless, it is very important to use rigorous criteria to identify a putative mutation as causative in order to avoid genetic “overdiagnosis” [10].
Although various genetic mutations have been associated with ACM they cannot account for the entire broad spectrum of disease expression. Therefore, epigenetic and environmental factors may act as disease modulators. For example, male predominance has been described as an independent risk factor for adverse outcomes. This may be related to increased serum testosterone levels in males [8]. Furthermore, it has been shown that genetic disease modifiers may account for disease severity. Most importantly, strenuous physical activity, particularly endurance sports, has been associated with early disease manifestation, disease severity and progression [11, 12].
Clinical phenotypes and symptoms
Right-dominant ARVC is considered the classical form of ACM, but left ventricular (LV) involvement is reported with a prevalence of up to 70%. Due to genetic heterogeneity, epigenetic and environmental factors there is a phenotypic continuum with right- and left-dominant subtypes at opposite ends. In right-dominant ARVC, a dilated RV with regional wall-motion abnormalities is observed. The subtricuspid region and right ventricular outflow tract (RVOT) are particularly prone to this remodelling process, leading to aneurysm formation. Biventricular arrhythmogenic cardiomyopathy is characterised by early involvement of both ventricles (figures 2 and 3). Disease progression is characterised by systolic impairment and biventricular dilation. In contrast to dilated cardiomyopathy, ventricular arrhythmias of both right bundle-branch block (RBBB) configuration – originating in the LV – and left bundle-branch block (LBBB) configuration are present at an early stage. Left-dominant arrhythmogenic cardiomyopathy (ALVC) has been proposed as a distinct form of ACM. It is characterised by the early occurrence of LV involvement (arrhythmias precede gross structural alterations), when global RV function is preserved [13]. Certain mutations are also associated to with LV dominant phenotypes, such as those involving DSP [14].
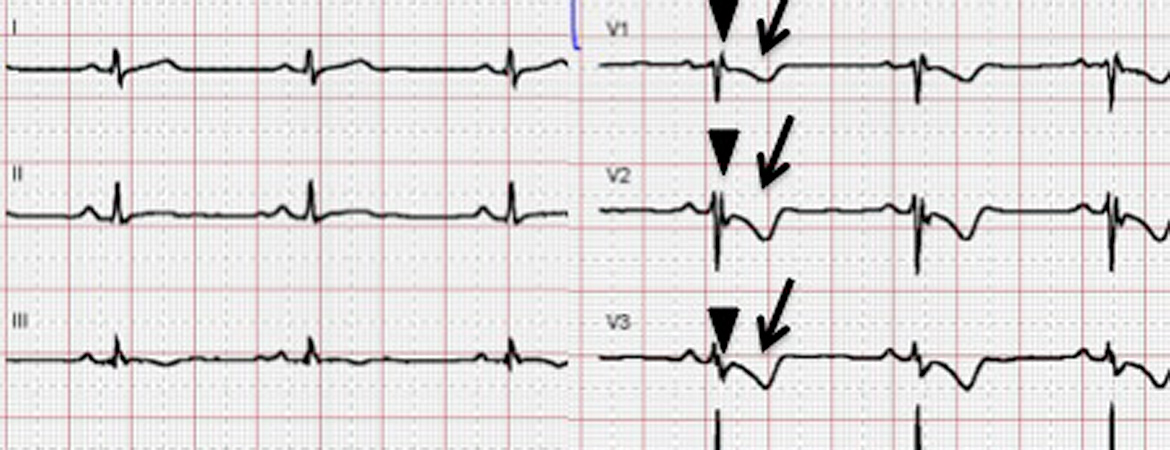
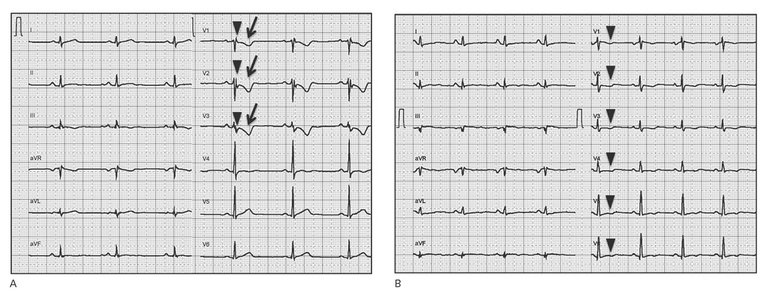
Figure 2: Electrical phenotypes of arrhythmogenic cardiomyopathy. A: Electrical phenotype of “classical” right ventricular arrhythmogenic cardiomyopathy. A 12-lead surface ECG (25 mm/s, 10 mm/mV) showing typical depolarisation abnormalities (prolonged terminal activation duration in V1-V3 ≥55 ms (arrowheads), a minor criterion according to the 2010 Task force criteria), and repolarisation abnormalities (T-wave inversions in V1-V3 in the absence of complete right bundle-branch block (RBBB) (arrows), a major criterion according to the 2010 Task Force Criteria in a patient with typical RV phenotype harbouring a plakophilin-2 mutation). B: Electrical phenotype of biventricular arrhythmogenic cardiomyopathy. A 12-lead surface ECG (25 mm/s, 10 mm/mV) showing repolarisation abnormalities (T-wave inversions in V1-V6 in the absence of complete RBBB (arrowheads), a major criterion according to the 2010 Task force criteria). T-wave inversions reaching V6 indicate biventricular involvement in this patient with biventricular ACM and a DSP mutation.
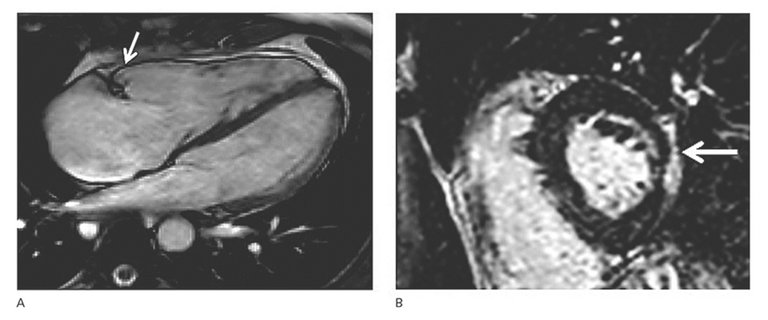
Figure 3: Structural phenotypes of arrhythmogenic cardiomyopathy.
A: Right ventricular (RV) dyskinesia in the RV subtricuspid area (arrow) during systole detected by cardiac magnetic resonance imaging (four-chamber view). The right ventricle is dilated (RV end-diastolic volume index 136 ml/m 2 , norm 65–102), and RV ejection fraction is reduced (RV-EF 38%, norm >45%). Regional RV dyskinesia in conjunction with RV dilation is considered a diagnostic criterion for classical ARVC diagnosis. Please also note that the right atrium is severely enlarged.
B: Epicardial left ventricular late gadolinium enhancement detected by cardiac magnetic resonance imaging indicating biventricular involvement in another patient with biventricular ACM (arrow).
ACM is a progressive disease. The course of the disease can be divided into four phases:
1. Concealed phase. Patients are asymptomatic without overt electrical or structural abnormalities.
2. Electrical phase. Symptomatic ventricular arrhythmias begin. SCD due to ventricular fibrillation (VF) can be the primary manifestation in this phase, which usually starts after puberty.
3. Structural phase. Ventricular dysfunction is present and heart failure symptoms may be present; this phase usually occurs in the third or fourth decade.
4. Phase of end-stage heart failure. This is usually the last phase of the disease and ~6% of all patients need to undergo heart transplantation. Nonetheless many patients do not develop this stage.
Of note, patients can go through so-called hot phases and superimposed myocarditis [25], in which an increased ventricular arrhythmia burden (e.g., electrical storm) can occur.
Diagnosis
Diagnosis can often be challenging and it is important to combine several diagnostic tools, including genetics whenever reasonable [11, 15]. A high clinical suspicion should be raised in athletes if symptoms correlate with premature ventricular beats or ventricular tachycardia (VT), particularly with LBBB morphology and a superior axis (II, III, aVF negative). However, LV forms or biventricular disease can also present with VT with RBBB morphology.
Generally, diagnostic criteria need to be reassessed during the disease course, since ACM is a progressive disease. Early diagnosis is particularly difficult, since many aspects of the disease may be absent. At this early stage, ACM can mimic other differential diagnosis such as idiopathic RVOT VT. We have previously reported that ECG criteria may change during the disease course, and repolarisation criteria in particular may be reversible. Furthermore, as a result of rapid changes in the genetic field, novel mutations can be sought, and mutations that were previously assigned as pathogenic may have turned into variants of unknown significance. Genetic counselling of probands and family members by cardiologists with expertise in cardiogenetics in collaboration with human geneticists is crucial to improve diagnosis. Therefore, it is important to re-evaluate diagnostic criteria in patients with ACM on a regular basis. In 2010, the original 1994 Task Force Criteria by McKenna et al. were revised by Marcus et al. to increase diagnostic sensitivity, particularly in affected, yet asymptomatic family members [16]. The clinician should bear in mind that the 2010 Task Force Criteria only apply to ARVC, but not left-dominant ACM.
The revised Task Force Criteria (table 1) consist of six diagnostic categories:
a) global/regional dysfunction and structural abnormalities of the RV;
b) histological characterisation;
c) repolarisation abnormalities on 12-lead surface ECG;
d) depolarisation abnormalities on 12-lead surface ECG;
e) ventricular arrhythmias;
f) family history and genetics.
A “definite” diagnosis can be made with two major criteria, one major and two minor criteria, or four minor criteria from different categories, a “borderline” diagnosis with one major and one minor criterion, or three minor criteria, and a “possible” diagnosis if one major criterion or two minor criteria are present.
Table 1: Revised 2010 Task force criteria, modified from Marcus et al. [16].
I. Global or regional dysfunction/structural alterations
Major
2D transthoracic echocardiography
• Regional RV akinesia, dyskinesia, or aneurysm •And 1 of the following criteria (end diastole): – Parasternal Long Axis (PLAX) RVOT ≥32 mm (PLAX/BSA ≥19 mm/m2) – Parasternal Short Axis (PSAX) RVOT ≥36 mm (PSAX/BSA ≥21 mm/m2) – or RV fractional area change ≤33%
Cardiac magnetic resonance tomography
• Regional RV akinesia, dyskinesia, or dyssynchronous RV contraction •And 1 of the following criteria (end diastole): – RV end-diastolic volume /BSA ≥110 ml/m2 (♂) or ≥100 ml/m2 (♀) – or RV ejection fraction ≤40%
RV angiography
• Regional RV akinesia, dyskinesia, or aneurysm
Minor
2D transthoracic echocardiography
• Regional RV akinesia, or dyskinesia •And 1 of the following criteria (end diastole): – PLAX RVOT ≥29–31mm (PLAX/BSA ≥16–18 mm/m2) – PSAX RVOT ≥32–35 mm (PSAX/BSA ≥18–20 mm/m2) – RV fractional area change >33 – ≤40%
Cardiac magnetic resonance tomography
• Regional RV akinesia, dyskinesia, or dyssynchronous RV contraction •And 1 of the following criteria (end diastolic): – RV end-diastolic volume/BSA ≥100–109 ml/m2 (♂) or ≥90–99 ml/m2 (♀) – or RV ejection fraction >40 – ≤45%
II. Histopathology (endomyocardial biopsy)
Major
Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium ≥1 sample, with or without fatty replacement
Minor
Residual myocytes 60–75% by morphometric analysis (or 50–65% if estimated), with fibrous replacement of the RV free wall ≥1 sample, with or without fatty replacement
III. Repolarisation abnormalities (>14 years of age)
Major
– T-wave inversions V1–V3 or beyond (in absence of complete RBBB)
Minor
– T-wave inversions V1–V2 or V4–V6 (in absence of complete RBBB) – T-wave inversions V1–V4, if complete RBBB present
IV. Depolarisation abnormalities
Major
Epsilon wave (reproducible low-amplitude signals between end of QRS complex to onset of the T-wave) in V1 to V3
Minor
Signal-averaged ECG (SAECG) with late potentials (if QRS on standard surface ECG <110 ms)
V. Arrhythmias
Major
– Non-sustained or sustained ventricular tachycardia (VT) of LBBB morphology with superior axis
Minor
– Non-sustained or sustained VT of LBBB morphology with inferior or unknown axis – >500 VES per 24 h (Holter)
VI. Family history
Major
– ARVC in a first-degree relative who meets current Task Force Criteria – ARVC confirmed pathologically at autopsy or surgery in a first-degree relative – Identification of a pathogenic mutation categorised as associated with ARVC/D in index patient
Minor
– Suspected ARVC in a first-degree relative (current Task Force criteria cannot be determined) – Premature SCD (<35 years of age) due to suspected ARVC in a first-degree relative – ARVC confirmed pathologically or by current Task Force Criteria in second-degree relatives
12-lead surface ECG, signal-averaged ECG (SAECG) and stress testing
The 12-lead surface ECG will be abnormal in up to 80% of patients. This will include T-wave inversions and terminal activation delay including epsilon waves in the right precordial leads V1–V3 (fig. 1). It should be kept in mind that the identification of epsilon waves may vary significantly among observers, and they should therefore be interpreted with caution, especially in the absence of other diagnostic criteria [17]. A SAECG can be helpful to identify delayed depolarisation in V1–V3.
Ventricular arrhythmias in ACM are often triggered by sympathetic activation. Thus, treadmill testing can reveal VT/VF or an increased premature ventricular contraction burden, sometimes with different morphologies [17].
Imaging: Transthoracic echocardiography (TTE), cardiac magnetic resonance tomography (CMR) and RV angiography
TTE is considered the initial imaging tool for suspected ACM and for screening family members. It may show RV/RVOT enlargement and regional contraction abnormalities, most commonly in the subtricuspid region and RVOT. We have recently suggested a novel RVOT measure to facilitate ACM diagnosis [18]. Novel technologies such as strain imaging allow for better quantification of regional wall motion, thereby allowing earlier detection of the disease and LV involvement. CMR has emerged as the non-invasive gold standard for assessing the RV. Late-gadolinium enhanced CMR can reveal myocardial fibrosis and fatty infiltration, but, because of a high intra-and inter-observer variability for assessing fibrosis and fat within the RV, this is not part of the diagnostic criteria (fig. 2). RV angiography is considered useful for diagnosis in experienced hands, owing to a positive predictive value of ~85%, and a negative predictive value of ~95%. However, as a result of its invasiveness, other diagnostic strategies are preferred [15].
Electrophysiological studies and electroanatomical voltage mapping
The goal of an electrophysiological study (EPS) in patients with ACM is the induction of sustained ventricular arrhythmias for diagnosis, risk stratification and to guide ablation. The susceptibility for arrhythmias, implantable cardioverter-defibrillator (ICD) treatment algorithms, and efficacy of antiarrhythmic drugs (AADs) can be assessed. Electroanatomic voltage mapping (EAM) is a technique that uses electrophysiology catheters to measure myocardial voltages. In diseased myocardium, lower voltages with a longer duration, splitting, and fractionation of signals can be recorded. Myocardial voltage maps can be obtained both from the endocardium and epicardium. EAM is generally safe, and it guides and improves outcomes of VT ablation. The diagnostic and prognostic utility of EAM has not been implemented in the current Task force Criteria, but recent data indicate that it can be useful for diagnosis and risk stratification [19].
Endomyocardial biopsy
Endomyocardial biopsy (EMB) has long been considered the diagnostic gold standard for ACM. Indeed, histological examination may allow confirmation of an ACM diagnosis. Biopsies are usually taken from the RV septum for safety reasons. Since the process of fibro-fatty infiltration usually spares the septum, EMB often yields false negative results. Nonetheless, EAM-guided biopsies taken from borderline diseased zones may improve diagnostic yield, and occasionally EMB can help to detect cardiac sarcoid, a common differential diagnosis of ACM.
Genetic testing
When performing genetic testing, rigorous criteria to ascertain a mutation as pathogenic should be used. Genetic testing is performed to confirm ACM in individuals with a high or intermediate (at least one major or two minor criteria) clinical suspicion, and to identify genetically affected relatives harbouring the pathogenic mutation of the index patient. A negative genetic test does not exclude ACM, since other unknown causal mutations and environmental factors may also cause the disease. The identification of pathogenic mutations may be useful in the differential diagnosis of ACM and phenocopies, such as myocarditis, idiopathic RVOT tachycardia, dilated cardiomyopathy (DCM), muscular dystrophies or sarcoidosis. Genetic cascade screening of relatives offers an alternative strategy to serial clinical evaluation. The absence in a family member of a clear pathogenic mutation causing the disease in the index patient obviates the need for serial clinical evaluation in this family member. Current consensus documents do not recommend genetic testing for risk stratification and therapeutic decision-making in ACM owing to insufficient data on genotype-phenotype correlation. It is crucial that genetic counselling is carried out by experienced personnel, as discussed in the diagnosis section, especially since pathogenicity of mutations may not always be clear. In particular, missense variants and variants of unknown significance should be interpreted with caution.
Differential diagnosis
A commondifferential diagnosis is idiopathic RVOT-VT, particularly at early stages of ACM without gross structural abnormalities. RVOT-VT is not associated with structural heart disease, and thus has a more benign course. The ECG during sinus rhythm is normal in idiopathic RVOT-VT. Speckle tracking echocardiography and ECG criteria during VT can help to differentiate both entities [20, 21]. Furthermore, idiopathic RVOT-VT responds well to verapamil, and an endocardial ablation is feasible and mostly curative, in contrast to ACM. The presence of multiple VT morphologies is very uncommon in idiopathic RVOT-VT, and suggests a structural heart disease such as ACM. Cardiac sarcoid can mimic ACM and should be considered if respiratory and systemic symptoms, high-grade atrioventricular conduction block and septal involvement are present, and familial disease is absent. Myocarditis can also mimic ACM and even sometimes overlap with it, since ACM can also lead to inflammation and lymphocytic infiltration [22]. Genetic mutations and a family history of cardiomyopathy are absent in classical myocarditis, whereas certain mutations (e.g. DSP) may predispose to ACM with superimposed myocarditis. Complete reversal of functional and structural changes suggests myocarditis. DCM is particularly difficult to distinguish from non-classic forms of ACM, and these two entities can significantly overlap and harbour similar mutations. Arrhythmias are mostly present in ACM at an early stage in the absence of large structural changes, unlike in DCM. Brugada syndrome can mimic ACM, but presence of structural abnormalities favours ACM. Other less common differential diagnoses include right ventricular infarction, congenital left-to-right shunts, Chagas disease, and Uhl’s disease. Finally, adaptation of the RV to increased workload in endurance athletes can mimic ACM and there is a debatable grey zone of what is considered physiological adaptation [23].
Therapy
The recently published international consensus statement on the treatment of ACM represents the latest recommendations [24]. Since ACM is a relatively rare disease, most recommendations rely on small cohorts and mostly observational studies. The restriction of physical activity, especially of endurance sports, is recommended in patients with a definite diagnosis of ARVC (class I), recreational low-intensity sports may be possible exceptions (class IIa). Pharmacological options consist of AADs (e.g., sotalol, amiodarone and mexiletine), beta-blockers, and heart failure drugs. AADs are recommended as an adjunct to ICD therapy in patients with frequent ICD discharges (class I). Moreover, they should be considered for improving symptoms due to frequent premature ventricular beats or non-sustained VT (class IIa), and may be considered as an adjunct to catheter ablation in selected patients with haemodynamically stable VT without an ICD (class IIb). Cardioselective beta-blockers should be considered in all patients with the ACM phenotype irrespective of arrhythmias (class IIa), but their prophylactic use in healthy gene carriers is not recommended. An ICD is indicated in patients who have experienced an episode of SCA or haemodynamically unstable, sustained VT (class I). If major risk factors such as unexplained syncope, moderate/severe ventricular dysfunction, or non-sustained VTs are present, ICD implantation should be considered. Nonetheless, there is no clear consensus regarding primary prevention of SCD in ACM patients without documented sustained VA/SCA. A single-chamber ICD is generally preferred in order to minimise the incidence of lead-related complications in this relatively young cohort. Since many patients benefit from antitachycardia pacing, subcutaneous ICDs play a limited role. Importantly, prophylactic ICD implantation is not recommended in asymptomatic patients without risk factors or healthy gene carriers (class III). Catheter ablation of VT is recommended in patients with incessant VT or frequent appropriate ICD interventions for VT despite drug therapy (on amiodarone: class I, off amiodarone: class IIa). If ≥1 endocardial ablation procedure fails, an epicardial approach is recommended (class I). The disease process in ACM begins in the subepicardial layers. Thus, in experienced hands, a combined endocardial/epicardial approach can be followed as an initial ablation strategy (class IIa). Since catheter ablation is not curative, it is not recommended as an alternative to ICD for prevention of SCD [24].
Future perspectives
The last two decades have substantially improved our understanding of ACM from a purely RV dysplasia to an inherited polygenic disease with a broad phenotypic spectrum potentially affecting both ventricles, being aggravated by environmental factors such as endurance sports. Emerging technologies in imaging, genetics, catheter ablation and device therapy have facilitated diagnosis, risk stratification and therapy of ACM, and prevention of SCD. Future challenges will involve early identification of asymptomatic patients and family members, improved risk stratification, and developing causal therapies to cure this challenging disease.
Key bullet points
– Arrhythmogenic cardiomyopathy (ACM) refers to a group of hereditary cardiomyopathies characterised by ventricular arrhythmias and structural/functional abnormalities of the ventricles.
– It is a common cause of sudden cardiac death in the young and in athletes.
– Causative mutations are mostly detected in desmosomal genes.
– Physical activity can promote disease onset, progression and arrhythmic events.
– Therapeutic strategies include restriction from sports, beta-blockers, implantable cardioverter-defibrillators, and catheter ablation.
Disclosure statement
No financial support and no other potential conflict of interest relevant to this article was reported.
Correspondence
Correspondence: Ardan M. Saguner, MD Department of Cardiology, University Heart Center Zurich Rämistrasse 100 CH-8091 Zurich ardan.saguner[at]usz.ch
References
1 Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–98.
2 Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–2.
3 Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–91.
4 Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al., (HRS) HRS and (EHRA) EHRA. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13:1077–109.
5 Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97:530–9.
6 Saguner AM, Duru F, Brunckhorst CB. Arrhythmogenic right ventricular cardiomyopathy: a challenging disease of the intercalated disc. Circulation. 2013;128:1381–6.
7 Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyö D, Rothenberg E, Delmar M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res. 2013;100:231–40.
8 Akdis D, Saguner AM, Shah K, Wei C, Medeiros-Domingo A, von Eckardstein A, Lüscher TF, et al. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: from a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur Heart J. 2017;38(19):1498–508.
9 Akdis D, Medeiros-Domingo A, Gaertner-Rommel A, Kast JI, Enseleit F, Bode P, et al. Myocardial expression profiles of candidate molecules in arrhythmogenic right ventricular cardiomyopathy/dysplasia compared with dilated cardiomyopathy and healthy controls. Heart Rhythm. 2016;13(3):731–41.
10 Medeiros-Domingo A, Saguner AM, Magyar I, Bahr A, Akdis D, Brunckhorst C, Duru F, Berger W. Arrhythmogenic right ventricular cardiomyopathy: implications of next-generation sequencing in appropriate diagnosis. Europace. 2016 May 18.
11 Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–55.
12 Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–806.
13 Saguner AM, Buchmann B, Wyler D, Manka R, Gotschy A, Medeiros-Domingo A, et al. Arrhythmogenic left ventricular cardiomyopathy: suspected by cardiac magnetic resonance imaging. Confirmed by identification of a novel plakophilin-2 variant. Circulation. 2015;132(6):e38–40.
15 Saguner AM, Vecchiati A, Baldinger SH, Rüeger S, Medeiros-Domingo A, Mueller-Burri AS, et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Cardiovasc Imaging. 2014;7:230–9.
16 Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–14.
17 Platonov PG, Calkins H, Hauer RN, Corrado D, Svendsen JH, Saguner AM, et al. High interobserver variability in the assessment of epsilon waves: implications for diagnostic of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2016;13:208–16.
18 Gotschy A, Saguner AM, Niemann M, Hamada S, Akdis D, Yoon JN, et al. Right ventricular outflow tract dimensions in arrhythmogenic right ventricular cardiomyopathy/dysplasia-a multicentre study comparing echocardiography and cardiovascular magnetic resonance. Eur Heart J Cardiovasc Imaging. 2017. May 26. Epub ahead of print.
19 Saguner AM, Medeiros-Domingo A, Schwyzer MA, On CJ, Haegeli LM, Wolber T, et al. Usefulness of inducible ventricular tachycardia to predict long-term adverse outcomes in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:250–7.
20 Saberniak J, Leren IS, Haland TF, Beitnes JO, Hopp E, Borgquist R, et al. Comparison of patients with early-phase arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract ventricular tachycardia. Eur Heart J Cardiovasc Imaging. 2017;18:62–9.
21 Hoffmayer KS, Machado ON, Marcus GM, Yang Y, Johnson CJ, Ermakov S, et al. Electrocardiographic comparison of ventricular arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2011;58:831–8.
22 Lopez-Ayala JM, Pastor-Quirante F, Gonzalez-Carrillo J, Lopez-Cuenca D, Sanchez-Munoz JJ, Oliva-Sandoval MJ Gimeno JR. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12:766–73.
23 Zaidi A, Sheikh N, Jongman JK, Gati S, Panoulas VF, Carr-White G, et al. Clinical Differentiation Between Physiological Remodeling and Arrhythmogenic Right Ventricular Cardiomyopathy in Athletes With Marked Electrocardiographic Repolarization Anomalies. J Am Coll Cardiol. 2015;65:2702–11.
24 Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36:3227–37.
25 Saguner AM, Roland F, Li GC, Fontaine GH. Superimposed myocarditis leading to heart transplantation in a young patient with arrhythmogenic right ventricular dysplasia. Eur Heart J. 2017;38:(Suppl. 1): ehx495.1933.